
来源:成都西南脑科医院在线咨询
肌张力障碍是临床常见的一种运动障碍性疾病,可发生在身体多个部位,依据肌张力障碍的发生部位,可分为局限性、节段性、偏身性和全身性。一般而言,发病年龄越早,症状可能越严重,波及身体其他部位的可能性也越大。近年来,我国肌张力障碍发病率呈逐年上升的趋势,对于这样一种疾病人们对它的发病原因与发病机制都充满了疑惑,下面将为大家简要介绍肌张力障碍发病原因与发病机制。
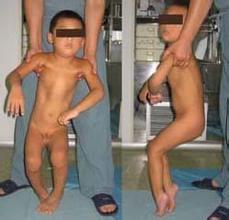
(一)肌张力障碍发病原因
目前,医学上将肌张力障碍分为原发性和继发性两型。所谓原发性也叫特发性肌张力障碍,主要是由于遗传因素而引起,60%以上的原发性肌张力障碍患者伴有家族史,剩余约40%患者为散发感染。
继发性肌张力障碍常是基底核,丘脑及脑干网状结构等病变的症候。如肝豆状核变性、核黄疸、神经节苷脂沉积症、苍白球黑质色素变性、进行性核上性眼肌麻 痹、双侧基底节钙化、甲状旁腺功能低下、中毒、脑血管病变、脑外伤、脑炎、脑裂畸形、药物诱发(L-DOPA、酚噻嗪类、丁酰苯类、胃复安、化疗药物)等 病因均可引发肌张力障碍。
(二)肌张力障碍发病机制
特发性肌张力障碍可为常染色体显性(30%~40%外显率),常染色体隐性或X连锁隐性遗传,显性遗传缺损基因DYT1已定位于9号常染色体长臂 9q32~34,编码ATP结合蛋白扭转蛋白A(torsin A),可有散发病例,环境因素如创伤或过劳等可诱发,如口-下颌肌张力障碍发病前,可有面部或牙损伤史,一侧肢体过劳,常诱发书写痉挛,打字员痉挛和运动员肢体痉挛等。
继发性肌张力障碍是纹状体,丘脑,蓝斑,脑干网状结构等病变所致,如肝豆状核变性,胆红素脑病,神经节苷脂沉积症,苍白球黑质红核色素变性,进行性核 上性麻痹,特发性基底核钙化,甲状旁腺功能低下,中毒,脑卒中,脑外伤,脑炎等;另外,药物(左旋多巴,吩噻嗪类,丁酰苯类,甲氧氯普胺)也可诱发。
病理及神经生化改变:特发性扭转痉挛可见非特异性病理改变,包括壳核,丘脑及尾状核小神经元变性,基底核脂质及脂色素增多,继发性扭转痉挛病理学特征随原发病不同而异,痉挛性斜颈,Meige综合征,书写痉挛和职业性痉挛等局限性肌张力障碍无特异性病理改变。
拮抗肌过度协同性收缩是本病主要的生理学特点,肢体每次收缩常由近端向远程扩展,肌电图检查发现,肌肉收缩间歇期无不自主运动电位,根据肌电图特点分为3型:
①持续30s的肌肉收缩,间隔短时间静息;
②重复,节律性收缩和静息状态,收缩期和静息期均为1~2秒钟;
③快速,短暂肌肉收缩,持续100ms,表现类似肌阵挛。
神经调控技术的诞生 有效解决肌张力障碍
近年来,肌张力障碍的发病率逐年上升,越来越多的家庭因此而辗转求医,受尽煎熬与折磨,更是有不少家庭为此而破裂!但值得庆幸的是,随着医疗技术发展,许多曾经不明原因造成的肌张力障碍已逐渐被攻克,“神经调控技术”的诞生,以其显著的稳定治治疗果、安全性高、治疗率高等优势深受广大肌张力障碍患者的信赖。
“神经调控技术”所采用的神经因子具有能够分裂增殖和向多种细胞分化的生物学特性及能力,病损部位的反射传导,将进入体内的神经因子方向趋化性增殖,来修复或替换损伤或坏死的肌细胞使坏死的肌纤维再生,从而恢复肌肉组织功能,进而恢复肌力和机体功能,达到肌张力障碍的临床治疗的目标。


![]()


地址:成都市武侯区武侯大道38号(二环高架武侯大道出口处)
电话:028-68760707








